MoleCode
Health Pass
- License — License: MIT
- Description — Repository has a description
- Active repo — Last push 0 days ago
- Community trust — 42 GitHub stars
Code Pass
- Code scan — Scanned 12 files during light audit, no dangerous patterns found
Permissions Pass
- Permissions — No dangerous permissions requested
No AI report is available for this listing yet.
Molecode presents molecules as code and enables LLMs to operate and reason on chemistry directly.
🧬 MoleCode
An LLM-native, graph-explicit molecular language
Official repository for MoleCode unlocks structural intelligence in large language models.
Molecode presents molecules as code and enables LLMs to operate and reason on chemistry directly.
Instead of making language models reconstruct molecular structure from cryptic strings,
MoleCode lets them read, write, and edit directly on the structures.
![]()

![]()
![]()
![]()
![]()
![]()
![]()
English | 中文
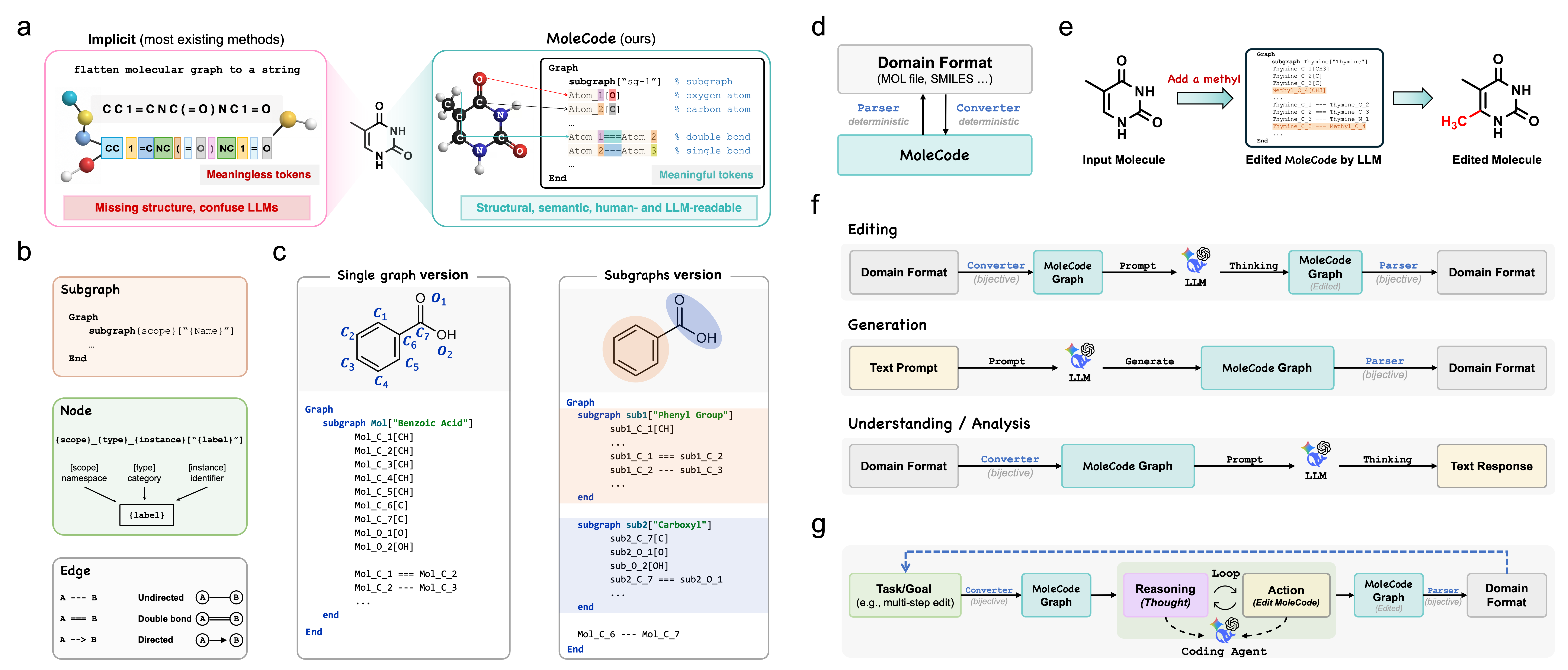
What is MoleCode?
A molecule is a graph: atoms are nodes, bonds are edges, and chemistry emerges from the topology. Yet large language models are almost always fed molecules as linear strings like SMILES, where the graph is implicit — connectivity is positional, branches are syntactic, and rings hide inside index digits. Before an LLM can do any chemistry, it must first reconstruct the graph from the syntax, spending reasoning budget on structural bookkeeping.
MoleCode makes the structure the language. Every atom and bond is written as a typed declaration with a persistent identifier, serialized as a Mermaid graph. Topology becomes directly readable, editable, and auditable inside the context window — and the format is deterministically and losslessly inter-convertible with SMILES / MOL via RDKit (no learned model, no information loss).
graph TB
subgraph chlorophenol["para-chlorophenol"]
chlorophenol_C_1[C]
chlorophenol_O_1[OH]
chlorophenol_C_2[CH]
chlorophenol_C_3[CH]
chlorophenol_C_4[C]
chlorophenol_Cl_1[Cl]
chlorophenol_C_5[CH]
chlorophenol_C_6[CH]
chlorophenol_C_1 === chlorophenol_C_2
chlorophenol_C_2 --- chlorophenol_C_3
chlorophenol_C_3 === chlorophenol_C_4
chlorophenol_C_4 --- chlorophenol_C_5
chlorophenol_C_5 === chlorophenol_C_6
chlorophenol_C_6 --- chlorophenol_C_1
chlorophenol_C_1 --- chlorophenol_O_1
chlorophenol_C_4 --- chlorophenol_Cl_1
end
The same
Subgraph → Node → Edgegrammar covers small molecules, polymers, and Markush structures — and extends to reaction mechanisms and multimodal document parsing.
Why it matters
| SMILES | MoleCode | |
|---|---|---|
| Topology | implicit, positional | explicit, named nodes & edges |
| Atom identity | none | persistent IDs (stable across prompt → reasoning → output) |
| Editing | whole-string rewrite | local graph op (add a methyl = 1 node + 1 edge) |
| Validation | fragile string parsing | deterministic RDKit round-trip |
| Reasoning behavior | memorizes syntax | generalizes over structure |
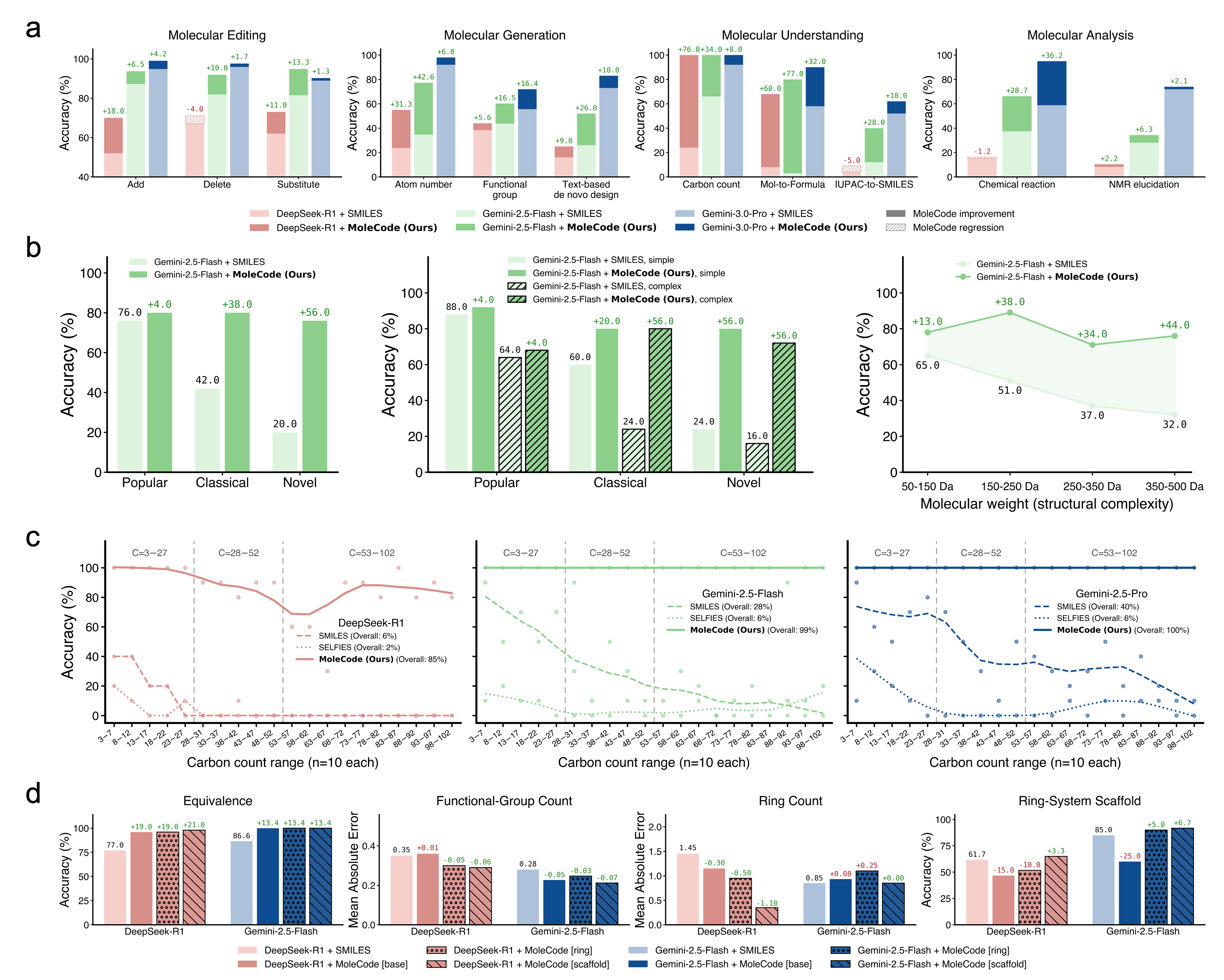
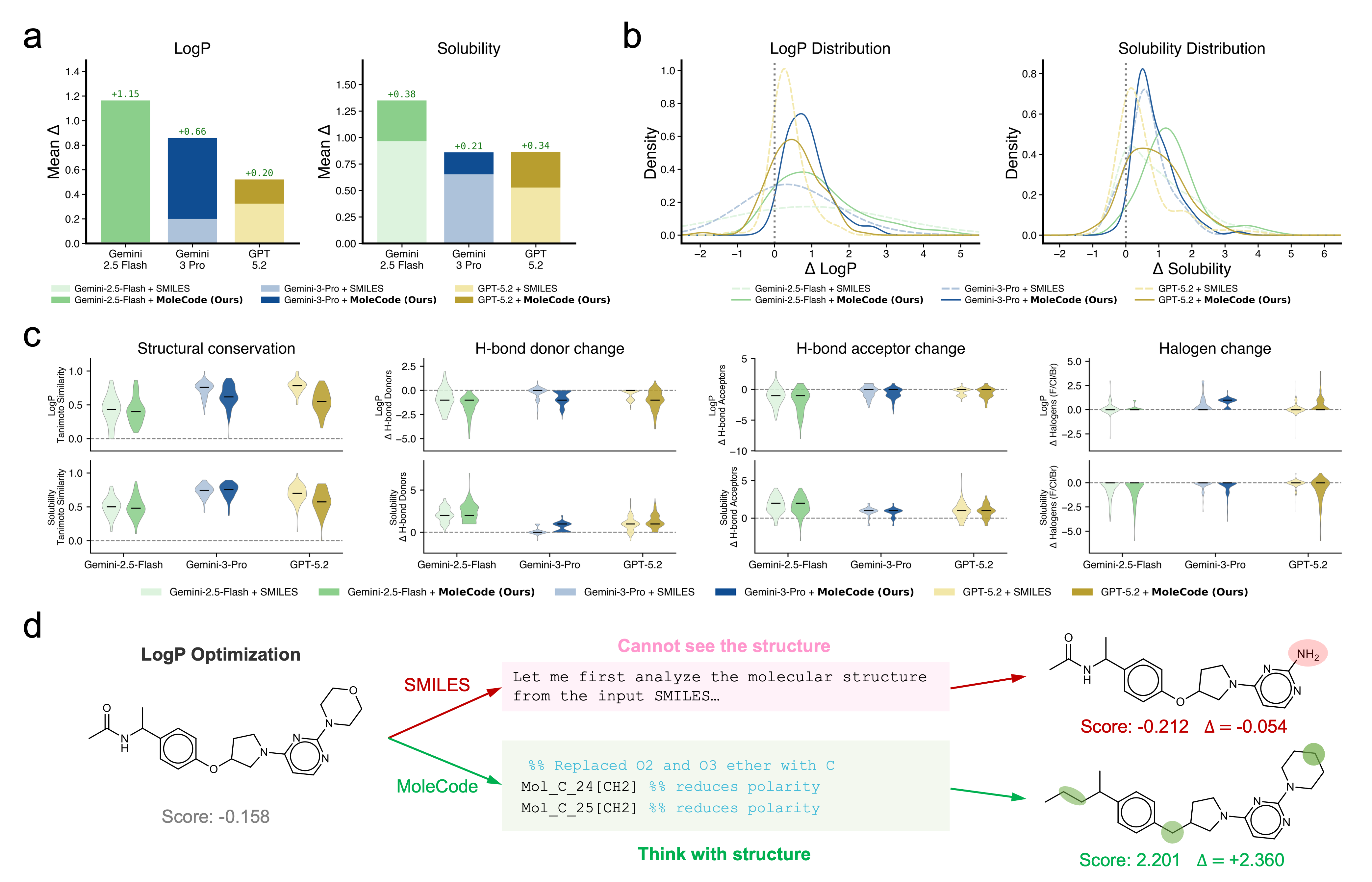
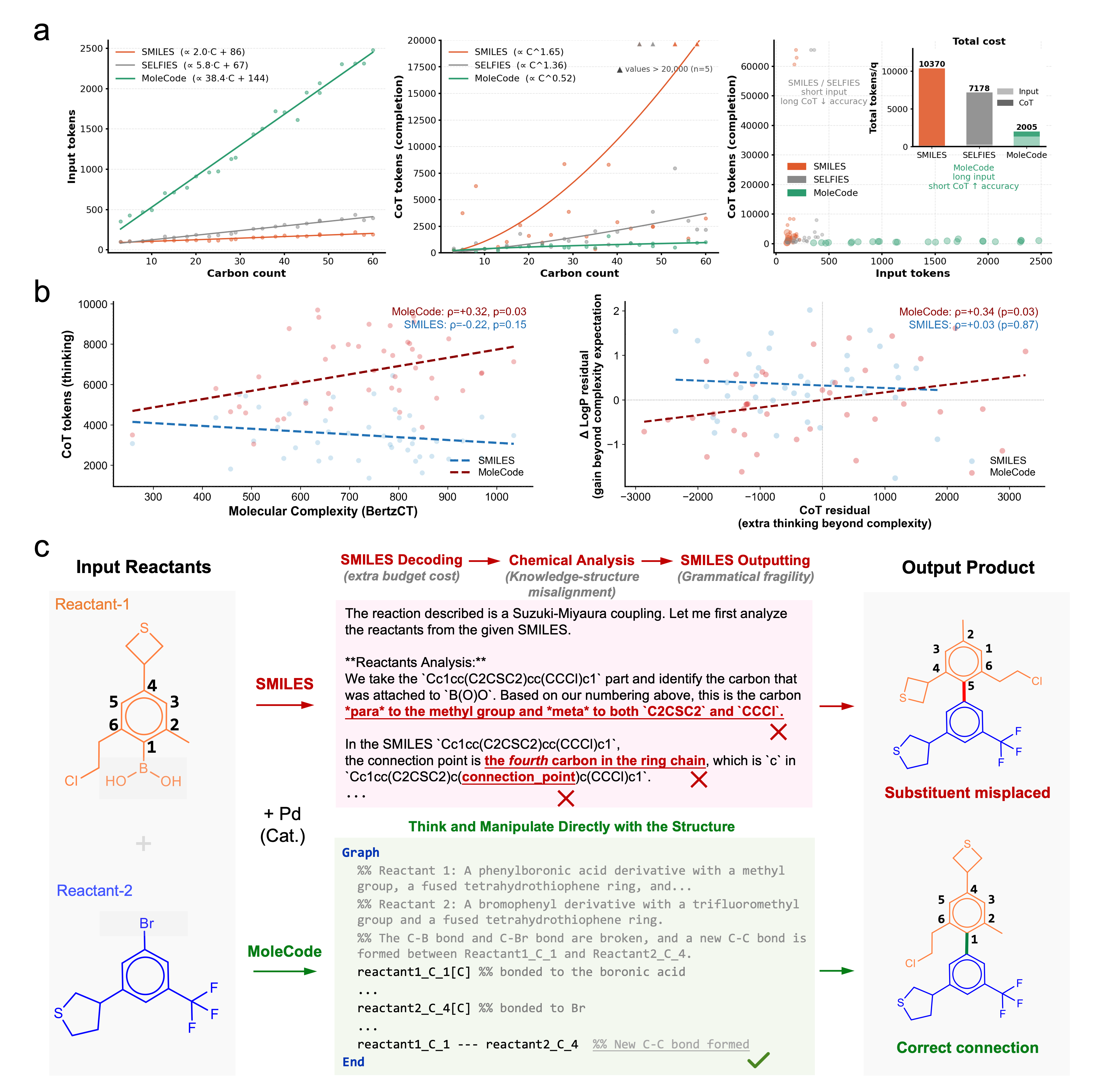
Empirically (see the MoleCode paper and docs/06-why-it-works.md):
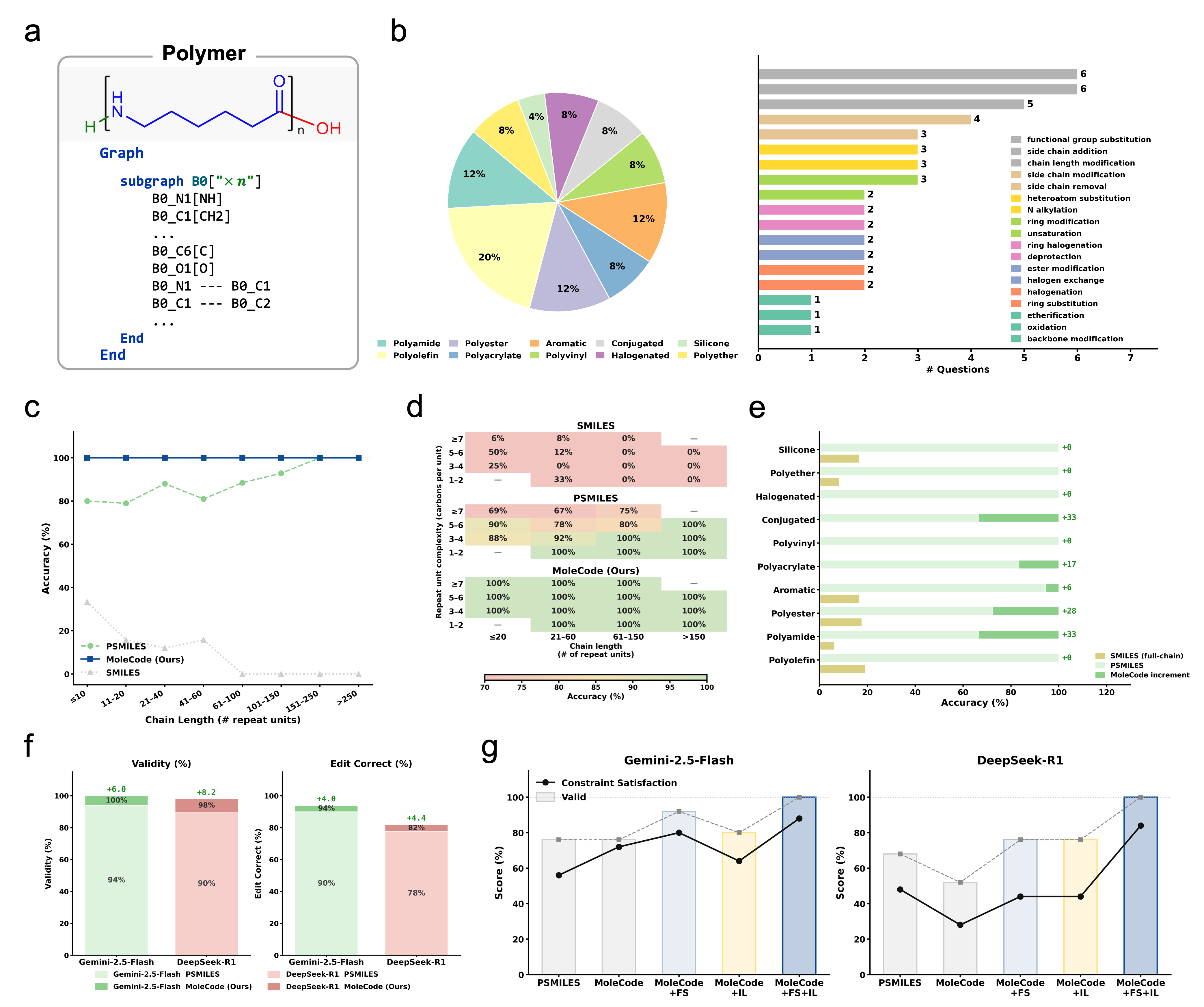
- Generalization, not memorization. SMILES accuracy collapses from ~42% on familiar molecules to ~20% on novel ones; MoleCode holds ~76–80% across all familiarity tiers.
- Cheaper reasoning. MoleCode has longer input but its chain-of-thought grows sub-linearly with molecule size (~C^0.52) versus SMILES' super-linear ~C^1.65 — about a 5× lower total token cost per query.
- Scales to big, repetitive objects. Full-chain SMILES accuracy falls toward 0% as polymer chains grow; MoleCode stays flat.
- Markush understanding jumps from 38.1% → 84.0%.
Install
git clone https://github.com/AtomFlow-AI/MoleCode.git
cd MoleCode
pip install -e . # installs rdkit + networkx
Quick start
from rdkit import Chem
from molecode import mol_to_mermaid, mermaid_to_mol, mol_to_smiles
# SMILES -> MoleCode graph
graph = mol_to_mermaid(Chem.MolFromSmiles("CC(=O)Oc1ccccc1C(=O)O"), name="Aspirin")
print(graph)
# MoleCode graph -> SMILES (lossless round-trip)
assert mol_to_smiles(mermaid_to_mol(graph)) == Chem.CanonSmiles("CC(=O)Oc1ccccc1C(=O)O")
Works with your coding agent (Claude Code · Codex)
MoleCode ships as a ready-to-use Agent Skill,
so coding agents can clone this repo and immediately reason over and edit
molecules at the explicit-graph level — no extra setup, no MCP server required.
| Agent | How it picks MoleCode up |
|---|---|
| Claude Code | Auto-discovers the skill at .claude/skills/molecode/. Just ask it to understand or edit a molecule. |
| Codex (and other agents) | Reads AGENTS.md at the repo root and uses the bundled CLI; interface metadata in agents/openai.yaml. |
Instead of asking the model to hand-write SMILES — error-prone for anything
non-trivial — the skill has it convert → inspect the named atoms/bonds → edit
the graph → validate, all through one stable CLI:
python .claude/skills/molecode/scripts/molecode_convert.py doctor
python .claude/skills/molecode/scripts/molecode_convert.py smiles-to-molecode "CCO" --name Ethanol
python .claude/skills/molecode/scripts/molecode_convert.py validate --input edited.mmd # formula, counts, round-trip
python .claude/skills/molecode/scripts/molecode_convert.py molecode-to-smiles --input edited.mmd
The skill bundles the six conversion forms (SMILES / PSMILES / Markush ↔
MoleCode) plus validate, compare (Markush-aware isomorphism) and doctor, a
syntax reference for hand-editing graphs, and a file-based edit workflow built
for large molecules. See .claude/skills/molecode/SKILL.md.
Three domains, one grammar
🧪 Small molecules — molecode.molecule
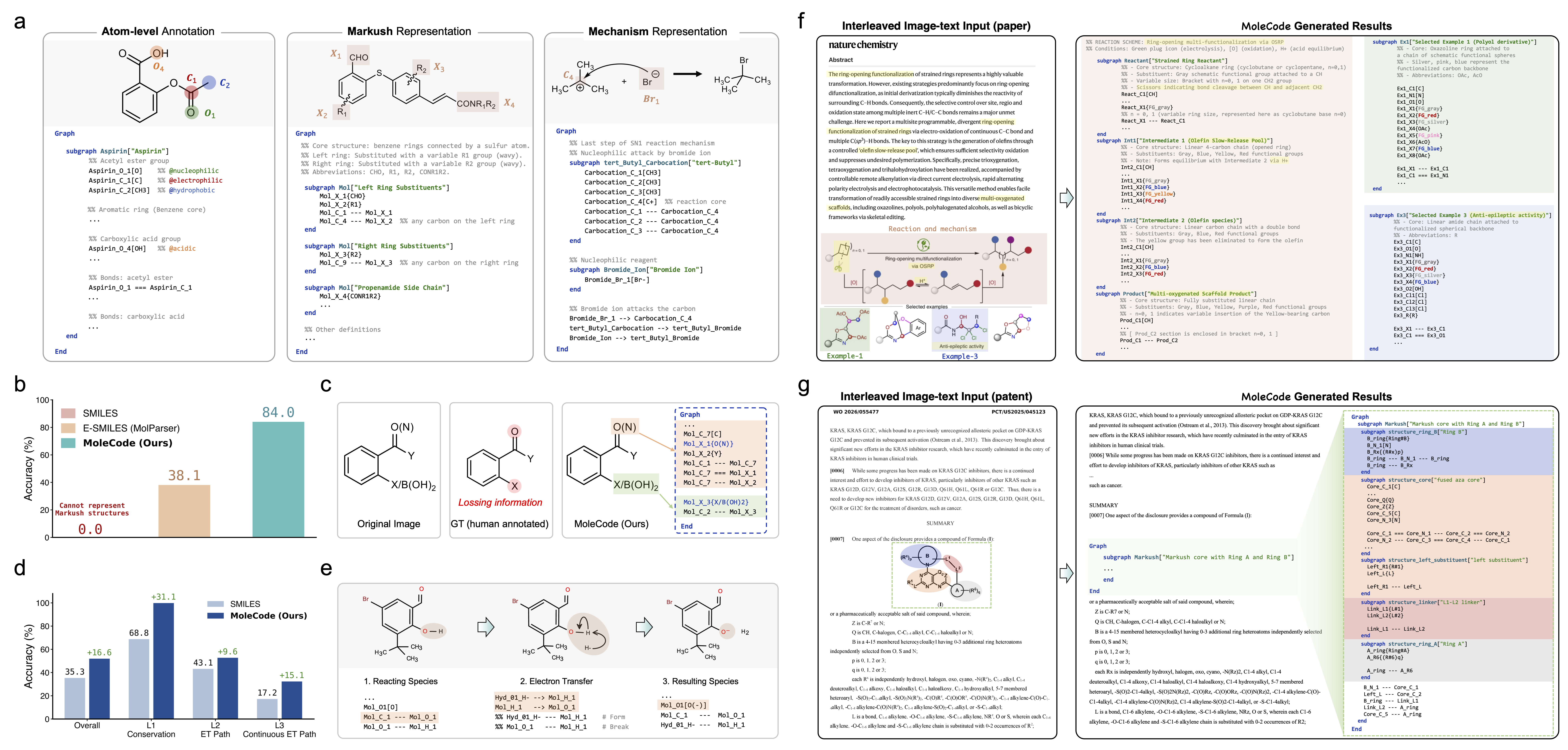
Atoms are prefix_Element_Number[Label] nodes; bonds are --- (single), === (double), -.- (triple), with ===|E|/===|Z| and _R/_S for stereochemistry. → syntax reference
🔗 Polymers — molecode.polymer
The repeat unit stays explicit as a subgraph carrying a symbolic ×n count, with TL/TR terminus markers — so the graph does not blow up with chain length. → polymer docs
from molecode.polymer import polymer_to_mermaid, mermaid_to_psmiles
graph = polymer_to_mermaid("*NCCCCCC(=O)*", n=8, name="Nylon-6") # PSMILES -> graph
mermaid_to_psmiles(graph) # -> '*NCCCCCC(=O)*'
🧩 Markush structures — molecode.markush
Variable R-groups and named substituents become abbreviation nodes in curly braces — {R1}, {Boc}, {Ar} — something plain SMILES cannot express. A built-in graph-isomorphism comparator scores predictions up to abbreviation expansion. → Markush docs
graph TB
subgraph Mol["molecule name"]
Mol_C_1[C]
Mol_O_1[OH]
Mol_X_1{Boc}
Mol_X_2{R1}
Mol_C_1 --- Mol_O_1
Mol_C_1 --- Mol_X_1
end
Run the tasks: understand · generate · edit · reason
MoleCode is a drop-in representation for any LLM — feed the grammar as a system prompt, hand the model a graph, and validate its output deterministically. The examples/ folder has runnable scripts for all four task families (they run offline by default, printing the exact prompt; set MOLECODE_API_KEY to call a model):
python examples/01_molecule_roundtrip.py # SMILES <-> graph (lossless)
python examples/02_polymer_roundtrip.py # polymers with ×n
python examples/03_markush_roundtrip.py # abbreviation nodes & isomorphism
python examples/04_understanding.py # count atoms / formula / rings ...
python examples/05_generation.py # de novo design under constraints
python examples/06_editing.py # local graph edits (add/del/substitute)
python examples/07_reasoning.py # reaction-product prediction
python examples/08_image_to_molecode.py # OCSR: molecule image -> MoleCode (vision model)
The reusable ingredients:
from molecode.prompts import MOLECULE_SYSTEM_PROMPT # give this to the LLM as the system prompt
from molecode.molecule import mol_to_mermaid # your molecule -> what the model reads
from molecode.molecule import mermaid_to_mol # model output -> validated RDKit Mol
Calling an LLM
MoleCode is just a representation, so you can use any LLM SDK — the prompts
are plain strings. For convenience the package also ships a tiny,
dependency-free, OpenAI-compatible client (molecode.llm.LLMClient, built on
stdlib urllib). You supply the API key and base URL — nothing is hard-coded, so
it works with OpenAI, DeepSeek, Azure, Together, vLLM, Ollama, …
from molecode import LLMClient
from molecode.prompts import MOLECULE_SYSTEM_PROMPT
from molecode.molecule import mol_to_mermaid, mermaid_to_mol
from rdkit import Chem
client = LLMClient(api_key="sk-...", base_url="https://api.openai.com/v1", model="")
# (or set MOLECODE_API_KEY / MOLECODE_BASE_URL / MOLECODE_MODEL and call LLMClient())
graph = mol_to_mermaid(Chem.MolFromSmiles("CC(=O)Oc1ccccc1C(=O)O"), name="Aspirin")
reply = client.chat(f"How many carbons are in this molecule?\n```mermaid\n{graph}\n```",
system=MOLECULE_SYSTEM_PROMPT)
print(reply)
Prefer the official openai SDK? Pass the same prompt strings straight toopenai.OpenAI().chat.completions.create(...) — you don't need LLMClient at all.
See docs/05-tasks.md for the full task catalog.
| Domain | Understanding | Generation | Editing | Reasoning |
|---|---|---|---|---|
| Molecules | ✅ | ✅ | ✅ | ✅ |
| Polymers | ✅ | ✅ | ✅ | — |
| Markush | ✅ | — | — | — |
Repository layout
molecode/ # the library (pip-installable)
├── molecule/ # small-molecule <-> Mermaid (rdkit_to_mermaid, mermaid_to_rdkit)
├── polymer/ # polymer <-> Mermaid (polymer_to_mermaid, mermaid_to_psmiles)
├── markush/ # Markush <-> Mermaid + graph isomorphism + abbreviation_map
├── prompts/ # LLM system prompts (molecule + markush grammars)
└── llm.py # optional OpenAI-compatible client (you supply key + base_url)
examples/ # 7 runnable demos (round-trips + 4 task families)
docs/ # overview, syntax, polymers, markush, tasks, why-it-works
AGENTS.md # entrypoint for coding agents
.claude/skills/molecode/ # Agent Skill: SKILL.md + CLI + references (Claude Code / Codex)
Results at a glance
| Generalization & reasoning | Goal-directed design | Scaling | Long molecules | General language |
|---|---|---|---|---|
 |
 |
 |
 |
 |
About AtomFlow
MoleCode is built and maintained by AtomFlow.
AtomFlow builds LLM-native AI for chemistry — letting language models operate
directly on molecular structure rather than on opaque strings. Our work centers on
molecule-grounded applications, including:
- Molecular chat & interaction — converse with a molecule; select atoms, bonds,
or fragments and edit them in natural language. - Structure-aware editing — auditable, graph-level molecular edits.
- Retrosynthesis — LLM-native synthesis planning over explicit structures.
- Literature reading & structure parsing — extracting structures from papers
and patents, including optical chemical structure recognition (OCSR).
MoleCode is the open representation layer underneath these products — making
molecular structure explicit, editable, and auditable for LLMs.
🌐 Learn more at atomflow-ai.com.
📚 Citation
If you use MoleCode in your research, please cite the MoleCode technical report:
@article{yan2026molecode,
title={MoleCode unlocks structural intelligence in large language models},
author={Yan, Zhiyuan and Liu, Chen and Zhao, Boxuan and Lin, Kaiqing and Zhao, Jixiang and Wang, Yimi and Lv, Liuzhenghao and Li, Hao and Zhang, Shanzhuo and Yuan, Li and others},
journal={arXiv preprint arXiv:2605.16480},
year={2026}
}
License
MIT © 2026 AtomFlow-AI
Reviews (0)
Sign in to leave a review.
Leave a reviewNo results found